-
靶向激肽B1受體通過AT1R依賴機制緩解高血壓
發布時間: 2025-08-29 點擊次數: 23次2025年8月21日,《Circulation Research》在線發表東卡羅來納大學Sriramula團隊的突破性成果。研究揭示,緩激肽B1受體(B1R)在中樞神經系統高血壓“神經炎癥爆發窗口"中扮演交感驅動主控角色:于Ang II輸注早期,B1R在下丘腦室旁核被選擇性上調,與AT1R直接組裝成功能復合體,驅動神經元超興奮、小膠質細胞激活及突觸前膜丟失,從而成為神經源性高血壓級聯的核心引擎。

背景:
神經源性高血壓以交感神經過度興奮為核心,中樞Ang II-AT1R通路被視為始動因素,但仍有部分患者對現有RAS抑制劑反應不佳,提示存在其他放大機制。緩激肽B1受體(B1R)在炎癥或損傷后迅速上調,可促炎、促交感,且前期研究發現其參與DOCA-鹽高血壓,但B1R是否及如何與中樞AT1R協同驅動Ang II型高血壓仍未知。本文即在這一空白背景下,系統探討B1R在高血壓中樞的作用及其與AT1R的相互作用。

結論1: 高血壓患者PVN中激肽B1R表達上調
研究發現,高血壓患者腦內B1R在PVN和SFO顯著上調,主要定位于神經元,并與血壓水平正相關。B1R與AT1R相互作用增強,提示其在中樞血壓調控中具有重要作用。
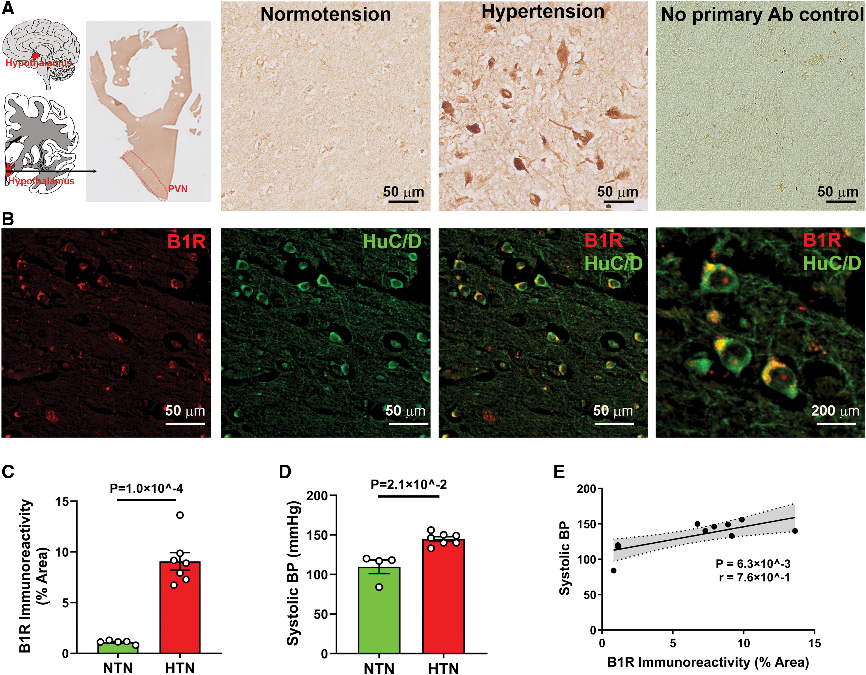
Fig1. 高血壓患者腦室旁核(PVN)神經元中B1R(激肽B1受體)
免疫反應性增加
結論2: 激肽B1受體在調控血壓的關鍵腦區中上調
Ang II誘導小鼠高血壓時,激肽-緩激肽系統被激活,B1R在下丘腦血壓調控核團顯著上調,尤以PVNzui突出。B1R主要定位于神經元和小膠質細胞。腦室注射B1R激動劑DABK可引起收縮壓升高,并在神經元中誘導B1R急性上調,證實B1R的中樞激活在高血壓病理生理中具有關鍵作用。
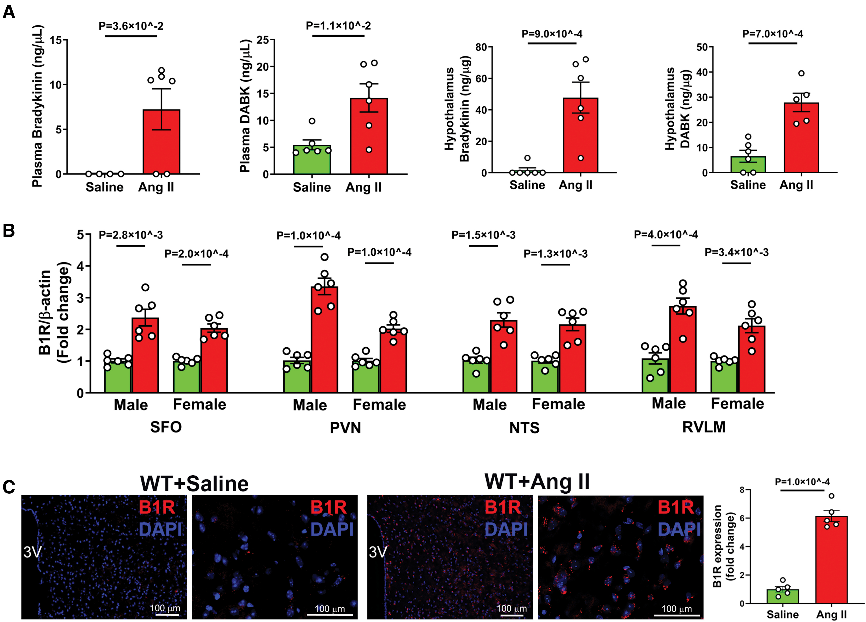
Fig2. 關鍵心血管調節中心中 B1R(激肽 B1 受體)表達增加
結論3:激肽B1受體缺失減輕神經源性高血壓及自主神經功能障礙
研究表明,B1R缺失可顯著減輕Ang II誘導的小鼠高血壓,維持壓力感受器反射功能,并抑制交感神經過度激活和體液調控異常。結果提示B1R在神經源性高血壓的發生發展及自主神經功能紊亂中發揮重要作用。

Fig3. 激肽B1受體(B1R)基因缺失可預防神經源性高血壓的發展
結論4: 阻斷B1受體減輕Ang II誘導的小膠質細胞活化及神經炎癥
研究表明,B1R在Ang II誘導的高血壓中促進小膠質細胞活化,導致促炎因子上調、抗炎信號下降,形成持續的神經炎癥微環境。該過程增強神經元活性和交感驅動,加重高血壓;而B1R缺失可顯著減輕炎癥反應和自主神經異常。

Fig4. B1R(激肽B1受體)阻斷可防止Ang II(血管緊張素II)誘導的
高血壓中的小膠質細胞激活
結論5:B1R的上調可誘導神經元過度活躍
B1R激活可直接增強皮層神經元放電頻率和爆發持續時間,模擬高血壓中交感驅動增強的特征;拮抗劑R715可顯著逆轉此效應。B1R對神經元同步性無影響,提示其通過增強單個神經元興奮性而非網絡整體協同來促進自主神經功能紊亂。

Fig5. B1R(激肽 B1 受體)刺激誘導神經元放電和爆發持續時間
結論6:B1受體定位于PVN突觸前位點并促使高血壓相關突觸丟失
研究表明,B1R在Ang II誘導高血壓中主要定位于突觸前終末,促進突觸前密度增加并導致突觸連接失衡,而非通過神經元凋亡介導。B1R缺失可減輕突觸結構異常,提示其在調控神經元興奮性、突觸傳遞及自主神經功能障礙中發揮關鍵作用。

Fig6. B1R(激肽 B1 受體)阻斷可減少 Ang II(血管緊張素 II)
誘導的突觸丟失
結論7:高血壓狀態下B1受體與AT1受體相互作用增強
研究表明,B1R與AT1R在PVN形成直接相互作用,Ang II可增強此受體復合物,促進神經源性高血壓。中樞B1R阻斷可減輕血壓升高和氧化應激,而外周阻斷無效。分子對接證實B1R-AT1R通過氫鍵和疏水相互作用形成復合物,DABK可結合其活性位點,揭示B1R-AT1R相互作用在高血壓中的分子機制。
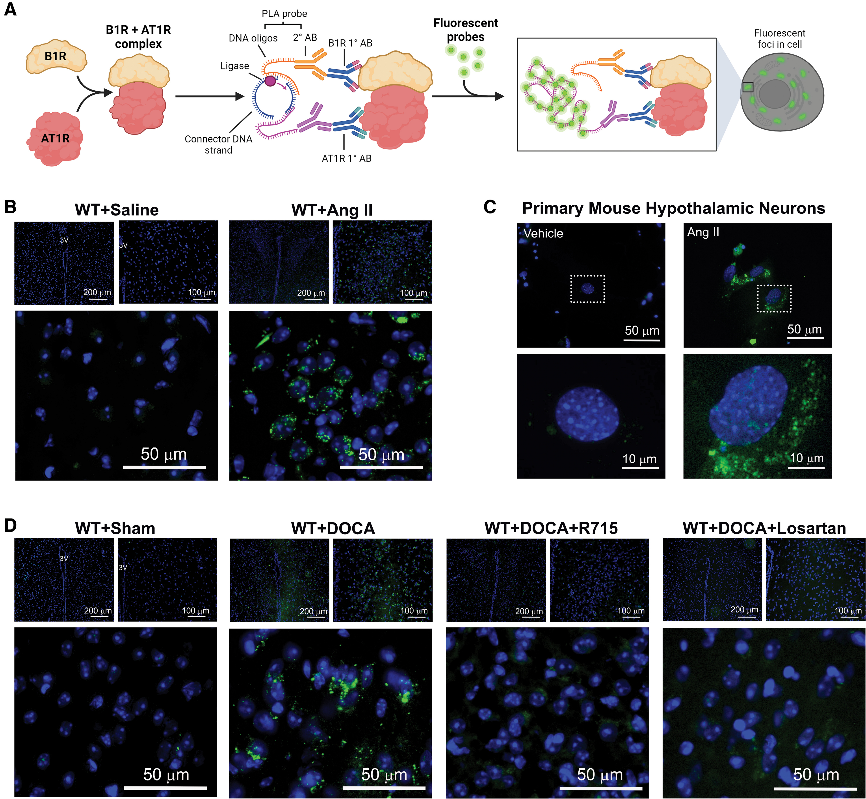
Fig7. AT1R-B1R(Ang II [血管緊張素 II] 1 型受體和激肽 B1 受體)之間的
相互作用在高血壓中上調
結論8:B1受體拮抗可減輕Ang II誘導的高血壓
SSR240612中樞阻斷B1R可有效降低Ang II誘導的血壓升高、減輕氧化應激并抑制PVN中B1R-AT1R相互作用。機制研究顯示,AT1R激活可上調B1R,B1R活化反向增強AT1R表達,形成雙向調控環路。這表明B1R在神經源性高血壓中通過調控受體互作和信號放大發揮核心作用。
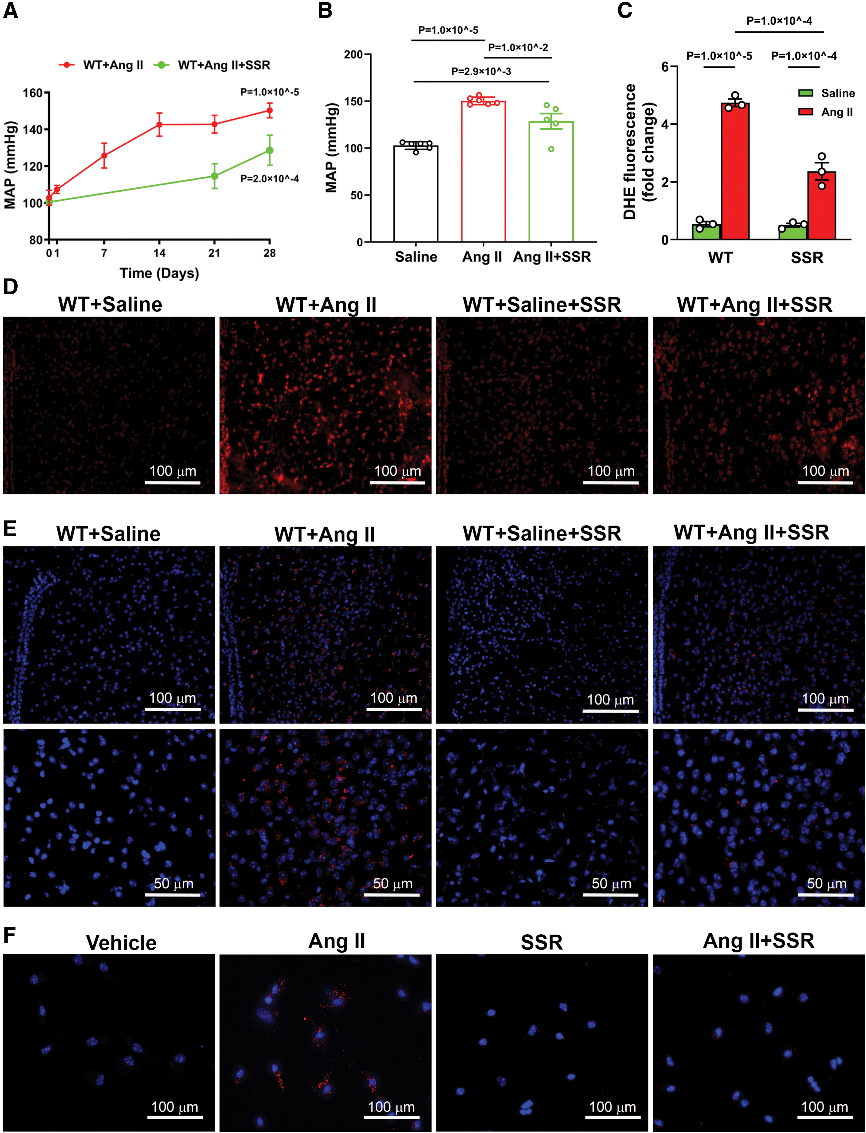
Fig8. B1R(激肽 B1 受體)的藥物阻斷可預防 Ang II(血管緊張素 II)誘導的高血壓
總結
本研究shou次在人高血壓腦中發現下丘腦室旁核B1R表達顯著升高并與血壓正相關;在小鼠體內證實B1R與AT1R直接結合,形成“Ang II→AT1R→B1R→放大交感輸出"的正反饋環路。基因敲除或藥理學阻斷B1R可顯著降低Ang II誘導的高血壓、恢復壓力反射、抑制小膠質細胞激活及神經炎癥,并防止突觸前膜丟失。超分辨定位顯示B1R主要位于谷an酸能神經元突觸前膜,MEA記錄證實其可迅速增加神經元放電。研究提出在現有ARB基礎上加用中樞B1R拮抗劑的新策略,但尚未驗證高血壓形成后的干預效果,且人體樣本量小,機制深度與長期安全性仍需進一步研究。



